The submission routes for Combination Products can be overwhelming. Read here the hack to the submission routes with the FDA for Drug-/Device-/Biologics Combination Products.
the basics
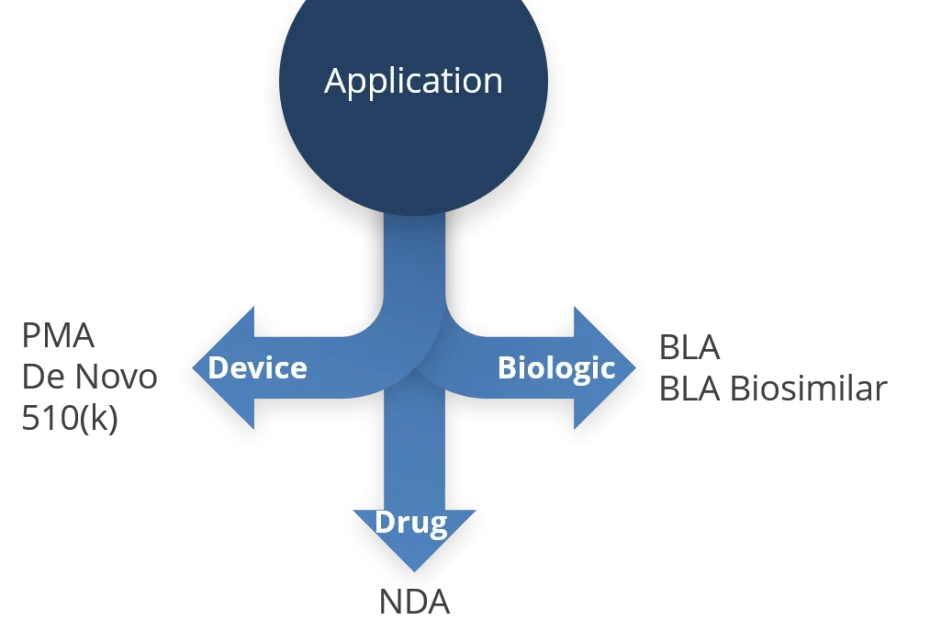
As it might already be known to the interested reader, any combination product is to be submitted to the Office of Combination Products (OCP) with the FDA. Whatsoever, there will be a lead office or lead center defined at point of submission. For this, the Primary Mode of Action (PMOA) is of utmost importance and in fact, decisive for the lead office.
For the ease of understanding, the possible lead offices shall be in the context of this blog the CDRH for devices, the CDER for drugs, and the CBER for biologics. Each of these centers can become the lead office, depending on the PMOA of the product to be submitted.
Device-led Combination Products
Device-led Combination Products (CPs) can be subject to either a Pre-Market Approval (PMA), a DeNovo Classification requrest or a 510(k) pathway.
PMA:
For any Class III device (or device-led CP), the PMA is mandatory with the FDA prior any product marketing. The FDA requires enough evidence to support the safe and effective use of the product. For CPs, this is to be demonstrated for all constituent parts of the CP. Important to say here, the PMA application requires technical data, non-clinical and clinical data, among other data and explanations.
De Novo:
The De Novo Classification request is a request to the FDA for device-led CPs, for which no classification has been established yet by the FDA and which should not be classified as Class III devices. With this, the De Novo route depicts a new type of device or device-led CP for which a Classification into class I or II is sought.
Be aware that the De Novo approach must be granted by the FDA and if the device or device-led CP falls into Class II, the product is understood as being able to legally market it based on safety and effectiveness data. With the classification into Class II, market authorization is granted subject to these general controls.
Such a product can serve later as a predicate to further Class II devices if substantial equivalence can be shown.
In the case that the De Novo Classification is not granted, the device or device-led CP falls into Class III for which a PMA is mandatory. Denial of a De Novo Classification can happen if the FDA thinks that general controls are insufficient for the product in question. This can stem from the drug- or biologic constituent part, for which special controls might be necessary.
510(k):
the Pre-Market Notification, also known as 510(k), is the route for which a manufacturer claims substantial equivalence to a previously marketed product with the FDA. Adhering to the strict rules to demonstrate equivalence, safety and effectiveness must be shown for such device-led CPs as well. Be aware that for a 510(k) substantial equivalence route, the underlying device (without the drug or biologic) cannot be taken as predicate, according to the FDA’s Guidance.
Drug-led combination product
For a drug-led CP, basically two approaches for receiving market authorization exist, i.e. the New Drug Application (NDA) and the Abbreviated New Drug Application (ANDA).
NDA:
New Drug Applications (NDA) are the generally valid approach to market approval for new drugs. Here, apart from generics and previously approved drugs, the Drug-led CP must demonstrate by documented evidence, the safety and effectiveness of the product (note here: PRODUCT, not constituents) for the prescription, recommendation or suggestion in the labeling.
Within the NDA approach, there are two types of NDA which can be followed, i.e. the stand-alone ( 505 (b)1) NDA, and the 505 (b)2 NDA. The difference between the two NDAs is in the fact that the stand-alone NDA holds full records of safety and effectiveness studies/data, conducted by the manufacturer or on behalf of the manufacturer, whereas a 505(b)2 contains or references third-party data.
ANDA:
Abbreviated New Drug Applications (ANDA) are the generally valid approach to market approval for drugs only in the case, in which the same active ingredient(s), dosage form, strength, route of administration, and conditions of use are present. This, in fact depicts a case, as an example, whereas a pre-filled syringe has been used and is newly used in an auto-injector.
To claim the ANDA route, safety and effectiveness must be shown to the “former” product as well as bio-equivalency to the reference listed drug (RLD). This is due to the fact, that the ANDA is relying on the safety and effectiveness of the RLD, instead of being required to collect data on the new product.
In any case, the constituent part(s) must be documented and underpinned with sufficient data to show compatibility for use with the final product’s formulation.
Biologic-led combination products
Very similar to the drug-led CP, for a biologic-led CP, basically two approaches for receiving market authorization exist, i.e. the Biologics License Application (BLA or stand-alone BLA) or via biosimilar or interchangeable BLA.
BLA:
Biologics License Application (BLA), is the route for biologics-led CPs for which all data for safety and effectiveness of the final product are demonstrated by the sponsor or on behalf of it. The stand-alone BLA is the route to choose when there is no biosimilarity or interchangeability claim possible with a previously marketed product by the FDA.
Biosimilar BLA:
In analogy to the ANDA-route, the Biosimilar BLA makes use of a previously cleared product by the FDA. For this benchmark product, biosimilarity or interchangeability can be claimed by the manufacturer. With this, the possibility is given to rely on previously demonstrated data on safety and effectiveness by the competitor or third party.
How Avanti Europe can help
Avanti Europe’s Experts have a decade-long track record and expertise in consulting and hands-on working in process implementation in the Pharmaceutical, Cosmetical, and Medical Device industry. Our experts support your company with hands-on workforce and support in risk-based process design, documentation, and training for the company staff. Visit our online shop for checklists and other services.